


Próbny II etap – rozwiązania cz. 2
Zapraszam na drugą część rozwiązań Próbnego II etapu czyli zadań z chemii organicznej.
PS Zbieram intensywnie na profesjonalny program do rysowania cząsteczek organicznych (owszem, istnieją darmowe, ale umożliwiają rysowanie pojedynczych cząsteczek, bez strzałek reakcji i reagentów, co jednak w syntezach się przydaje + oczywiście wiele innych bajerów). Tak czy inaczej, podczas naszego wspólnego przygotowania do kolejnej, 66. edycji Olimpiady Chemicznej będzie to już wyglądało dużo lepiej i profesjonalniej.
Jeszcze raz przypominam o ankiecie (ANKIETA), która ma dwa cele :
- Stworzyć solidne statystyki, aby móc realnie porównać się do innych.
- Ma dać mi opinię na temat zadań w samych sobie – czy zostały dobrze przygotowane, czy dobrze przygotowują do OlChemu itd, ponieważ chcę to wszystko nieustannie ulepszać i za rok zrobić taką edycję, że będzie już nie do odróżnienia od tej prawdziwej!
Moje przemyślenia na temat zadań :
- Zadanie 4 – stereochemiczne łamigłówki : jest to zadanie wskazywane na jedno z najłatwiejszych. Generalnie, wpasowuje się ono w tematykę folderu, chociaż w ogóle uważam, że nie musiałaby się pojawiać żadna taka przesłanka w FW, aby takie zadanie mogło się pojawić. Pierwsze reakcje są dość elementarne i często spotykane w poprzednich edycjach. Połączenie odrobiny prostego NMR’a , ale na którym i tak nie opierało się zadanie, w przeciwieństwie do następnego zadania z tą syntezą. Generalnie poszło Wam dobrze, raczej nic nie sprawiło problemu. Poszło zatem zgodnie z planem, ponieważ moim zdaniem było to najprostsze zadanie z całej piątki.
- Zadanie 5 – synteza alkaloidu : jest to najtrudniejsze Waszym zdaniem z tego próbnego II etapu. Punktem zaporowym zdecydowanie druga linijka syntezy, a więc przejście ze związku D do F z reakcją intramolekularną oraz izomeryzacją. A nawet jeśli ktoś przeszedł ten etap, to dalej też nie było wcale łatwiej. To zadanie miało w sobie wszystko, co trudne i nie ma się co dziwić, że sprawiało problemy.
Zadanie 4 – rozwiązanie
a) To zupełnie rutynowe odświeżenie podstawowych reakcji, w której należy zwrócić dodatkowo uwagę na stereochemiczne aspekty – czy byłoby to zasygnalizowane w FW czy też nie. Oczywiście w tym roku mamy całe zadanie dotyczące stereochemii, zatem to się pojawiło.
Oto reakcja cykloheksenu z nadmanganianem potasu w środowisku zasadowym – zwróćcie uwagę, że produkt przejściowy (jego rysowanie nie było wymagane) tłumaczy stereochemię produktu czyli związku A.

Powstały związek faktycznie jest mezo, co wynika z tego, że jest on symetryczny.
Warto nadmienić, że alternatywnie zamiast nadmanganianu (koniecznie w odpowiednim środowisku, bo przecież środowisko kwaśne spowoduje efekt podobny do ozonolizy utleniającej) można użyć \(1.OsO_{4} \ 2. NaHSO_{3} \)
Reakcja z dowolnym nadkwasem (zapisanym jako \(RCO_{3}H \) jest poniżej. Jako modelowego przykładu użyłem tutaj mCPBA, aby przy okazji przypomnieć Wam ten bardzo ważny reagent.

Zauważcie, że wystarczyło znać tylko jedną z reakcji i wtedy po informacji z zadania (że A oraz A’ to stereoizomery) dało się od razu napisać strukturę drugiego. Epoksydy w zdecydowanej większości przypadków ulegają otwarciu w taki sposób, że powstałe dwie grupy są po przeciwnych stronach, zatem struktura związku A’ :

Związki A oraz A’ to para diastereoizomerów.
b) związek A jako związek mezo nie będzie skręcał płaszczyzny światła spolaryzowanego (związek optycznie nieaktywny).
c) Teraz związki C, D oraz E. Reakcja tworzenia związku C to powtórka reakcji, która już była przed chwilą, pojawia się tylko nowe pytanie – jaka będzie wzajemna relacja przestrzenna pomiędzy nowo wytworzonym atomem tlenu w epoksydzie oraz grupą \(-OH \) ? Zanim odpowiemy na to pytanie, napiszmy jeszcze reakcję naszego wyjściowego związku z chlorkiem acetylu, co jest reakcją doskonale Wam znaną i nie wymaga ona żadnego rozjaśnienia. Zobaczcie, że powstały ester nie jest w stanie być dawcą protonu do wiązania wodorowego, o którym mowa w tekście, podczas gdy nasz wyjściowy związek ma grupę hydroksylową, a więc tym dawcą może być. Z tego wnioskujemy, że w tworzeniu związku C chodzi o te wiązania wodorowe, podczas gdy na tworzenie związku E wpływają czynniki steryczne (i faktycznie, mamy przecież dużą grupę \(-OCOCH_{3} \)).
Zdecydowanie łatwiej wykombinować związek E, ponieważ domyślamy się, że powstały epoksyd będzie uciekał od rozpychającej się grupy (po prostu – szukamy takiego rozwiązania, w którym podstawniki są jak najdalej od siebie, a więc dążymy do jak najmniejszego zatłoczenia sterycznego). Zatem ten estrowy podstawnik oraz grupa epoksydowa muszą być po przeciwnych stronach pierścienia. I w takim razie w związku C pewnie chodzi o produkt, w którym oba podstawniki są po tej samej stronie. Aby to wiązanie wodorowe w produkcie przejściowym się wytworzyło, to potrzebujemy aby tlen z epoksydu był jak najbliżej grupy hydroksylowej.

Na powyższym schemacie mamy, idąc odpowiednio od lewej strony : związek C , (cykloheks-2-enol), związek D oraz związek E.
d) Schemat syntezy DET :
Swoją drogą, jak już jesteśmy przy okazji kwasy but-2-enodiowego, to chciałem tylko odświeżyć dwa kwasy, które często pojawiają się na II czy III etapach, a mianowicie :
Kwas maleinowy (czyli izomer cis) : 
Kwas fumarowy (czyli izomer trans) : 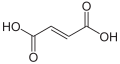
Pytają czesto o to, że kwas maleinowy może ulegać odwodnieniu do odpowiedniego związku cyklicznego, podczas gdy dla kwasu fumarowego jest to niemożliwe, ponieważ te grupy karboksylowe są za daleko od siebie – fajna metoda na rozróżnienie obu tych izomerów.
\(HOOCCH=CHCOOH \xrightarrow{RCO_{3}H} \) 
\(\xrightarrow{1. NaOH, \ H_{2}O \ 2. EtOH, \ H^{+}}\) 
*W samym schemacie nie trzeba było zaznaczać żadnej stereochemii.
Projekcja Fischera dla formy R,S :
$
e) Nie istnieje żadna korelacja pomiędzy kierunkiem skręcania płaszczyzny światła spolaryzowanego a konfiguracją na danym centrum asymetrii. Przewidywanie \(( \pm ) \) na podstawie konfiguracji R,S jest równie skuteczne co wróżenie z fusów.
f) Wzór naszego katalizatora to \(M(OR)_{x} \) , a więc być może jest to alkoholan jakiegoś metalu. Można do tego zadania podejść bardziej w sposób obliczeniowy (sami wiecie, jak wiele da się wyczarować z zawartości procentowej, która moim zdaniem jest najpotężniejszą daną jaką możecie dostać, niczym prezent) albo bardziej na podstawie podanej informacji o widmie \(^{1}H \ NMR \)
Pojawienie się dwóch sygnałów o intensywnościach względnych 6 : 1 , co moglibyśmy jeszcze zapisać z uwzględnieniem odpowiednich multipletowości : (6H, d) oraz (1H, spt) to absolutny spektroskopowy klasyk, który powinniście już co najmniej kilka razy widzieć i umieć go zinterpretować jako grupę izopropylową. Mamy w niej element symetrii, przez co dwie grupy metylowe są równocenne i stąd mamy (6H) , obok których jest grupa \(-CH \) , zatem korzystając z reguły \(n + 1 \) przewidujemy, że grupy metylowe wystąpią w postaci dubletu. Analogicznie, grupa \(-CH \) wystąpi jako septet (czasem też już się to zapisuje jako multiplet, tutaj jednak w zadaniu musiałem to bardziej doprecyzować)
[Komentarz] : nie ma, wg mojej skromnej wiedzy, ustalonego skrótu dla septetu (ja użyłem spt) , więc traktujcie to jako zapis brudnopisowy. Bo gdyby pojawiło się na zawodach widmo w formie rysunku, to ja bym sobie do brudnopisu ,,przepisał” je właśnie w formie (ileś atomóm H, rodzaj sygnału : singlet (s) , dublet (d) itd. ) i byłoby to do użytku mojego własnego.
Nasz katalizator można by już przepisać w formie : \(M(OCH(CH_{3})_{2})_{x} \)
A więc masa tego katalizatora to \(M_{M} + 59x \) , a masa samego węgla wynosi \(36x \) , gdzie \(M_{M} \) to masa molowa pierwiastka z bloku d (jak informuje treść zadania).
Wiadomo jeszcze, że zawartość procentowa węgla wynosi \(50,7 \% \) . Mogę z tego wyznaczyć masę molową całego katalizatora (\(M \)) czy też masę molową metalu :
\(0,507 = \frac{36x}{M} = \frac{36x}{M_{M} + 59x} \implies M_{M} = 12x \)
Zobaczcie, że nawet nie ma sensu sprawdzanie dla \(x \leq 3 \) bo wtedy wychodzi masa mniejsza niż skandu, czyli pierwszego pierwiastka z bloku d.
Dla \(x = 4 \) otrzymujemy \(M_{M} = 48 \approx 47,87 \implies M = Ti \)
W takim razie wzór katalizatora to : \(Ti(OCH(CH_{3})_{2})_{4} \)
Tutaj jego struktura :

[Czy moglibyśmy mieć zatem chiralne, organiczne związki tytanu?]
Jego otrzymywanie jest proste, można bowiem to zrobić z chlorku tytanu (IV) :
\(TiCl_{4} + 4 (CH_{3})_{2}CHOH \rightarrow Ti(OCH(CH_{3})_{2})_{4} + 4HCl \)
g) Pojawienie się reakcji Sharplessa na Olimpiadzie to kwestia czasu. Jest to reakcja, za którą posypała się nagroda Nobla i w zasadzie dla wielu jest pierwszą reakcją, która przychodzi do głowy na myśl o reakcji, w której ważna jest jeszcze stereochemia.
Jest to reakcja, która na pewno robi niezłe wrażenie i sprawia, że dane zadanie automatycznie wydaje się trudne, a potem okazuje się zupełnie przeciwnie. Pamiętajcie, że reakcję wytłumaczoną możecie dostać naprawdę każdą, nawet absolutnie najtrudniejszą (przykładowo, nic nie stoi na przeszkodzie, aby pojawiła się reakcja Dakina-Westa) , zatem też mija się z celem uczenie się wszystkich reakcji – wystarczą te z Murry’ego, serio.
[Komentarz] : reakcja Sharplessa to dla mnie must-know. Ja bym się jej nauczył (z wyjątkiem rozróżniania \(\pm \) DET , czyli z której strony powstanie ten epoksyd., bo to na pewno musiałoby być podane. Ale sam schemat reakcji (czyli czego używamy, oraz co z czego powstaje) to bym znał.
Ja idąc na II etap z organy czułem się zdecydowanie najlepiej, a podczas pierwszych minut na zawodach omal nie dostałem zawału, kiedy w zadaniu 5 [ 59 edycja, II etap, Zadanie 5 ]zobaczyłem katalizator rutenowy i nieznaną mi reakcję. Okazało się, że to zadanie było banalne i do zrobienia na czysto w 25 minut. Więc jak zawsze powtarzam – pierwsze wrażenie to sobie można wsadzić do… kosza!
Zacznijmy od narysowania (Z) – tridec-2-en-1-olu. Kto pomylił się w rysowaniu tej cząsteczki ma powody do zmartwień. Pamiętajcie, żeby odświeżyć sobie nazewnictwo i rysowanie na podstawie nazwy, chociaż po dwa przykłady dla każdej grupy funkcyjnej i może kilka związków, które zawierają kilka grup funkcyjnych. Ostatnio częściej pojawiają się pytania z nomenklatury.

Zobaczcie, że powyższy związek faktycznie należy do alkoholi allilowych, tak jak mamy wspomniane w treści zadania przy opisywania reakcji Sharplessa.
Patrząc na podany schemat reakcji Sharplessa, jeszcze nie uwzględniając kwestii stereochemicznych widzimy, że generalnie jest to reakcja epoksydowania, więc po prostu mamy przejście \(alken \rightarrow epoksyd \)
W naszym przykładzie używamy stereoizomeru \((-)-DET \) , a więc to dolny schemat w przedstawionym rysunku w treści nas będzie interesował. Teraz wystarczy narysować sobie cały ten nasz ,,tridecen” tak samo jak to w modelowym schemacie i analogicznie dołączyć atom tlenu. Uważam, że to naprawdę nic trudnego i punktacja aż 5 punktów to swojego rodzaju nagroda za dotrwanie do końca zadania.
Poniżej schemat reakcji (użyłem już skrótowego zapisu ,,R” dla całej tej reszty alkilowej). Pamiętajcie, że jeżeli robicie sobie takie skróty, to warto w czystopisie napisać co oznacza dane R, a nie że jakaś samowolka). Nasz wyjściowy związek ma konfigurację Z i tak też trzeba go narysować. Centra konfiguracji mają przypisaną stereochemię.

Zadanie 5 – rozwiązanie
a) Zaczynamy zatem zadanie od ustalenia struktury związku A. Zbierzmy informacje, które mamy dostępne :
- pik \((M+1) ^{+} \) jest dla \(m \slash z = 103 \) skąd odczytujemy, że masa związku A wynosi \(M_{A} = 102 \ \frac{g}{mol} \)
- w widmie \(^{13}C \ NMR \) mamy pięć pików, zatem jest co najmniej 5 atomów węgla w związku A. Jeden z sygnałów występuje przy około 175 ppm, co sugeruje obecność grupy karbonylowej \(C=O \)
- w widmie \(^{1}H \ NMR \) mamy sygnały o następujących intensywnościach względnych \(1 : 0,64 : 0,66 : 0,96 \) co po sprowadzeniu do najmniejszych wartości całkowitych daje \(\approx 3 : 2 : 2 : 3 \) , a to sumarycznie daje nam 10 atomów wodoru (lub każdą wielokrotność, jednak raczej będzie to 10 atomów, biorąc pod uwagę masę molową związku A)
- związek A zawiera grupę karbonylową oraz węgiel \(\gamma \) , co sugeruje obecność fragmentu \((\gamma) C-C-C-C=O \) . Tej informacji dostarcza nam treść zadania dotycząca związku A’ , bo tam tylko atom jodu się zmienia (w miejsce wodoru gamma), więc w związku A taki fragment musi właśnie być.
Jak widzimy, informacji jest aż nadto. Zakładając wzór ogólny dla związku A : \(A = C_{x}H_{y}O_{z} \) możemy napisać :
\(M_{A} = 102 = 12x + y + 16 z \)
Sprawdźmy czy dla \(z = 1 \) oraz \(y = 10 \) wyjdzie jakaś całkowita liczba atomów węgla (bo jeden tlen na pewno będzie od grupy karbonylowej, a dziesięć wodorów mamy ze spektroskopii).
\(102 = 12x + 10 + 16 \implies x = 6,33 \) , a więc jest źle.
Spróbujmy zatem dwa atomy tlenu, wodory zostają takie same :
\(102 = 12x + 10 + 32 \implies x = 5 \)
Zatem związek A to \(C_{5}H_{10}O_{2} \)
Idąc za Claydenem, stopień nienasycenia oznaczymy sobie jako DBE. Ja tak robię w brudnopisie zawsze, bo pisanie słownie ,,stopień nienasycenia” zajmuje dużo czasu. A jaki Wy macie na to skrót to bez różnicy 🙂
Dla związku A mamy \(DBE = 1 \) co sugeruje jeden pierścień lub jedno wiązanie podwójne. Wiedząc jednak, że jest tam obecna grupa karbonylowa, to wiemy, że jest to wiązanie podwójne między węglem a tlenem \(C=O \) i nie będzie tam żadnych pierścieni.
*b) Przyczyną pojawienia się piku, który jest o jeden większy od piku jonu molekularnego jest obecność izotopu \(^{13}C \)
c) Podsumujmy sygnały z \(^{1}H \ NMR \) z uwzględnieniem rozszczepienia każdego sygnału.
\((3H,s) \) , \((2H,sxt) \) , \((2H,t) \) , \((3H,t) \) – pochodzą one odpowiednio od grup \(-CH_{3} \) , \(-CH_{2} \) , \(-CH_{2} \) , \(-CH_{3} \)
Zwykle najłatwiej interpretować singlety, ponieważ takie grupy wodorów (tutaj musi to być \(-CH_{3} \)) nie mogą sąsiadować z innymi wodorami bezpośrednio (konkretnie w ,,odległości” trzech wiązań). W takim razie jedynym możliwym sąsiadem jest albo pozbawiona atomów wodoru grupa karbonylowa albo bezpośrednio atom tlenu. Biorąc pod uwagę wartość przesunięcia tego sygnału \(\approx 3,7 \ ppm \) moglibyśmy właściwie jednoznacznie postawić na fragment \(-OCH_{3} \)
Ze względu na brak innych singletów wnioskujemy, że od razu obok tej grupy metoksylowej musi stać grupa karbonylowa czyli łącznie dając ester metylowy \(RCOOCH_{3} \)
Dalsza część to banał, ponieważ mamy juz ustalony fragment \((\gamma) C-C-C-C=O \) co daje tylko jedną możliwość dla związku A : \(CH_{3}CH_{2}CH_{2}COOCH_{3} \)
Pozostaje przypisanie sygnałów (nie po kolei, uszeregowane wg łatwości przypisania sygnałów) \(^{1}H \ NMR \) :
- \(\delta_{1} \approx 3,7 \ ppm \implies -OCH_{3} \)
- \(\delta_{2} \approx 2,3 \ ppm \implies -CH_{2}C=O \)
- \(\delta_{4} \approx 0,95 \ ppm \implies CH_{3} \) (chodzi wodory gamma)
- \(\delta_{3} \approx 1,65 \ ppm \implies -CH_{2}- \) (chodzi o wodory beta)
W widmie \(^{13}C \ NMR \) :
- \(\delta_{1} \approx 175 \ ppm \implies C=O \)
- \(\delta_{2} \approx 50 \ ppm \implies -OCH_{3} \)
- \(\delta_{3} \approx 38 \ ppm \implies -CH_{2}C=O \) (chodzi o węgiel alfa)
d) Ustalenie A’ to wręcz nagroda za ustalenie związku A, nic trudnego. \(A’ = ICH_{2}CH_{2}CH_{2}COOCH_{3} \)
Schemat syntezy (zapiszę go dwa razy, bo pisząc cały związek robi się troche niewyraźnie, więc pierwszy zapis jest całościowy, w drugim używam skrótu R dla reszty, która jest niereaktywna w danej reakcji) :
\((A’ )\ ICH_{2}CH_{2}CH_{2}COOCH_{3} \xrightarrow{CH_{3}CH_{2}OH} (B) \ ICH_{2}CH_{2}CH_{2}COOCH_{2}CH_{3} \)
\(ICH_{2}CH_{2}CH_{2}COOCH_{2}CH_{3} \xrightarrow{NHBn_{2}} (C) \ Bn_{2}NCH_{2}CH_{2}CH_{2}COOCH_{2}CH_{3} \)
\(Bn_{2}NCH_{2}CH_{2}CH_{2}COOCH_{2}CH_{3} \xrightarrow{(COOEt)_{2} \ , \ NaOEt} (D) \ Bn_{2}NCH_{2}CH_{2}(COOEt)CH([O=C]COOCH_{2}CH_{3}) \)
Dla czytelności wstawiam jeszcze wzory wszystkich produktów :
- \(A’ = ICH_{2}CH_{2}CH_{2}COOCH_{3} \)
- \(B = ICH_{2}CH_{2}CH_{2}COOCH_{2}CH_{3} \)
- \(C = Bn_{2}NCH_{2}CH_{2}CH_{2}COOCH_{2}CH_{3} \)
- \(D = Bn_{2}NCH_{2}CH_{2}(COOEt)CH([O=C]COOCH_{2}CH_{3}) \)
Przejście \(A’ \rightarrow B \) : \(RCOOCH_{3} \xrightarrow{CH_{3}CH_{2}OH} RCOOCH_{2}CH_{3} \)
Przejście \(B \rightarrow C \) : \(ICH_{2}R \xrightarrow{NHBn_{2}} Bn_{2}NCH_{2}R \)
Przejście \(C \rightarrow D \) : \(RCH_{2}COOEt \xrightarrow{(COOEt)_{2} \ , \ NaOEt} R(COOEt)CH([O=C]COOCH_{2}CH_{3}) \)
Pierwsza reakcja to ciekawa reakcja o nazwie transestryfikacja, która polega na wymianie reszty alkoholowej w estrze na drugą, czyli w tym przypadku wymieniamy resztę metoksylową na etoksylową – powstaje związek B. Następnie dodajemy aminę, z wplątanym tam zabezpieczeniem w postaci grup benzylowych (typowa grupa zabezpieczająca dla grup \(-OH /-NH \) ). Jest to Elementarna reakcja substytucji nukleofilowej, również nie powinna ona sprawiać problemu i tak też tworzy się związek C. Tworzenie związku D to sugerowana w folderze wstępnym reakcja kondensacji (tutaj konkretnie reakcja Claisena, a więc kondensacja pomiędzy dwoma estrami), w której użyty nad strzałką reagent (szczawian dietylu) nie jest w stanie wytworzyć jonu enolanowego, zatem to związek C jest jedynym możliwym wyborem na jego utworzenie. Jako, że jest to ester to nie mamy dylematu, z której strony stworzy się jon enolanowy, zatem struktura produktu jest jednoznaczna. Podany wzór sumaryczny związku D jednocześnie określa czy odeszła cząsteczka wody czy też nie.
e) hydroliza związku A’ to oczywiście hydroliza estru, zatem powstanie kwas. Następcze dodanie wodorowęglanu sodu spowoduje deprotonoację tego kwasu, a powstały nukleofil obecny w grupie \(O=C-O^{-} \) zaatakuje węgiel związany z jodem \(( \delta +) C-I ( \delta-) \) co skutkuje reakcją wewnątrzcząsteczkową.
Powstanie związek Y : 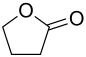 , a obecna w nim grupa funkcyjna to oczywiście lakton.
, a obecna w nim grupa funkcyjna to oczywiście lakton.
Przy okazji chciałbym pokazać fantastycznie nadającą się na II etap reakcję jodolaktonizacji :

f) gdyby w reakcji zamiast szczawianu dietylu użyć estru kwasu malonowego, to rodzi się wstępnie problem, ponieważ zarówno kwas malonowy może ulegać enolizacji jak i ester. Jeśli jednak weźmiemy pod uwagę obecność drugiej formy rezonansowej dla enolu kwasu malonowego to można powiedzieć, że jest on stabilniejszy i jego tworzenie będzie faworyzowane w porównaniu do enolu estru, bez stabilizujących form rezonansowych.
Poniżej formy rezonansowe malonianiu dietylu po deprotonacji \(NaOEt \)
[Komentarz] : moim osobistym zdaniem, w dobrym tonie leży rozpisanie takich form rezonansowych podczas odpowiadania na takie pytanie. Jeśli ktoś jeszcze czuje się w tym temacie niepewnie, to zapraszam tutaj : Rezonans – nauczmy się tego!

Biorąc pod uwagę powyższe rozważania, rysujemy wzór związku Z :
\(RCH_{2}COOCH_{2}CH_{3} \xrightarrow{CH_{2}(COOEt)_{2} , \ NaOEt} RCH_{2}C(=O)CH(COOEt)_{2} \)
*znów używam skrótu R, ponieważ jak widzieliście wyżej, dla tak długich związków robi się po prostu nieczytelnie.
g) Oto wzory porduktów wraz z wyjaśnieniem poniżej :
\(Bn_{2}NCH_{2}R \xrightarrow{H_{2} , Pd/C} (E) \ H_{2}NCH_{2}R \)



Przejście \(D \rightarrow E \) to typowa reakcja odbezpieczenia grupy aminowej, o czym już wspominaliśmy, że została ona już dodana w formie zabezpieczonej, podczas tworzenia związku C. Według mnie, powinniście tą reakcję po prostu znać – pojawia się ona nawet w folderze wstępnym części A z identyczną podpowiedzią, jaką tutaj podałem w treści : 62 edycja – A4 (tutaj to już nawet w kosmicznym wydaniu, bo jako rozcinanie estru, co nawet na finał się nadaje, jednak efekt można powiedzieć ten sam – ,,rozcinamy” grupę benzylową).
Pomimo iż dokładny mechanizm jest dość złożony, to jednak dość prosto to wszystko się przedstawia jeśli chodzi o sam efekt, bo przecież cząsteczkę wodoru (\(H-H \)) zwyczajnie przyłączamy w miejsce benzylu, dodając na jego końcu jeden atom wodoru i mając w efekcie toluen. Rozcięcie w tym miejscu powoduje deficyt jednego wiązania przy azocie, więc naturalnie uzupełniamy go drugim atomem wodoru.
Następnie przejście do związku E’ to reakcja intramolekularna (tzn. wewnątrzcząsteczkowa). Szukamy w naszym związku grup, które mogłyby ze sobą reagować. Widzimy grupę aminową (aminową dodatkowo sugeruje uprzednie jej odbezpieczenie, zatem logiczne, że pewnie teraz chcemy, by nam reagowała) oraz ketonową i estrową. Reakcja aminy z grupą estrową wiązałaby się z odejściem etanolu, co jest niemożliwe wg dalszych warunków zadania, bo wiemy, że hydroliza związku H daje 3 mole etanolu, a tak to by dała 2 mole. Zatem grupy estrowe muszą pozostać nienaruszone. Pozostaje możliwość keton + amina (drugorzędowa) co skutkuje powstaniem iminy – związek E’.
Izomeryzacja do związku F to trudna moim zdaniem reakcja, jednak tak skonstruowałem to zadanie, aby ta reakcja nie miała znaczenia dla dalszej części zadania, ponieważ następcza reakcja z wodorem i tak usunie wiązanie podwójne. Reakcja ta jest reakcją izomeryzacji i polega na przesunięciu wiązania podwójnego z iminy na C=C , co jest faworyzowane (siła napędowa reakcji) ponieważ powstały produkt (związek F) ma trwalszy, potrójny układ sprzężenia (\(O=C-C=C-C=O \)) , który zaznaczyłem jeszcze kółkiem.
h) Wzory produktów wraz z wyjaśnieniem poniżej :




Tworzenie związku H to reakcja aza-Michaela. Było to wyjaśnione w tekście, ponieważ tego typu reakcja jest bardziej charakterystyczna dla finału niż II etapu (chociaż też może się pojawić, zwłaszcza patrząc na tegoroczny FW), ale dlatego też była tutaj wytłumaczona. Mamy informację, że jest to addycja sprzężona, a jako że związek G nie jest związkiem sprzężonym, to wiemy, że ta addycja będzie miała miejsce na wiązaniu C=C w estrze, który dodajemy nad strzałką i w takim razie amina ze związku G będzie pełniła rolę nukleofila (co też jest powiedziane).
Następna reakcja to ponownie kondensacja pomiędzy estrami (Claisena), ale tutaj pojawia się problem, który z estrów będzie ulegał enolizacji i taką też podpowiedź znajdujemy w treści zadania – będzie to ester, który jest najłatwiej dostępny ze względów sterycznych. Zatem będzie to ten ester, który przed chwilą dołączył się w reakcji aza-Micheala.
Kolejny etap, czyli tworzenie związku J to typowa sekwencja, którą spotykamy podczas syntez z użyciem kondensacji itp. Mamy bowiem najpierw reakcję hydrolizy (rozszczepia się ester, pamiętajmy o tym, że oba estry ulegają rozszczepieniu!) i potem reakcję dekarboksylacji, która jest charakterystyczna dla wzajemnego ułożenia dwóch grup karbonylowych, które też zaznaczyłem kółkiem na rysunku związku I (tzw. \(1,3 – diCO\) ) , z czego jedna musi być grupą karboksylową i tak też mamy u nas.
Potem mamy reakcję z elementarnym odczynnikiem, do którego absolutnie żadnych wskazówek nawet nie wolno Wam podawać, bo należy wiedzieć, że jest to odczynnik selektywny i zredukuje nam grupę ketonową do alkoholu, nie naruszając przy tym kwasu karboksylowego. W razie czego odsyłam tutaj : Dwa najważniejsze reduktory : LiAlH4 oraz NaBH4 . Reakcja ma w sobie dodatkowy haczyk, bo wiemy, że związek K ma być tricykliczny, a póki co mamy dwa pierścienie, zatem w tej reakcji musi tworzyć się dodatkowy pierścień. Jedyną opcją jest tutaj reakcja powstałej grupy \(-OH \) z kwasem karboksylowym tworząc ester, a konkretnie lakton.
i) Zmiana masy molowej o 4 g podczas tworzenia związku P najprawdopodobniej jest wynikiem dodania czterech atomów wodoru, a zatem reakcją redukcji. Dodatkowo, wiemy, że związek P jest bicykliczny, a więc w tym przejściu musimy mieć jeszcze rozerwanie pierścienia, a więc nietrudno znaleźć nasze centrum, na którym ma się odbyć reakcja. Skoro ma być to redukcja estru (laktonu) to w sumie jedynym odczynnikiem przychodzącym na myśl jest \(LiAlH_{4} \) (reagent X) , który zredukuje ester do dwóch kwasów i rozerwie pierścień, a dodatkowo powstały produkt ma 3 centra stereogeniczne co oznacza obecność ośmiu możliwych stereoizomerów, co spełnia wszystkie podane w zadaniu warunki.








Dodane komentarze (10)
Czy ta grupa funkcyjna w laktonie to na pewno lakton? Czy to nie jest tak, że ten związek zaliczany jest do laktonów, a grupa w nim obecna to po prostu grupa estrowa? Z góry dziękuje za wszystkie odpowiedzi.
Jeśli ktoś w odpowiedzi napisał ,,cykliczny ester” to powinien mieć komplet punktów, bo jakby nie patrzeć jest to prawda (chociaż przykładowo nie wolno amidu nazwać ,,ketoaminą” ). Moim zdaniem to odrębna grupa funkcyjna, ponieważ nazewnictwo laktonów różni się od nazewnictwa zwykłych estrów.