


Mechanizmy w kinetyce chemicznej – cz.1
Zgodnie z Waszą prośbą, wrzucam posta, w którym postaram się trochę przybliżyć mechanizmy w kinetyce chemicznej.
Fakt, przesłanka, że może to być dla Was, zawodników idących za chwilę na II etap (ale to szybko mija!) ważne jest to, że ten element (zupełnie niezapowiedziany!) pojawił się teraz na I etapie. Dla mnie to ewidentna wskazówka, że mogą pytać o mechanizmy – najbardziej prawdopodobne, że będzie to miało związkiem z teorią (przybliżeniem) stanu stacjonarnego albo z rysowaniem bądź układaniem mechanizmu reakcji. W związku z tym, że teoria stanu stacjonarnego jest generalnie prostym zagadnieniem i do tego jest świetnie opisana w Atkinsie, to zajmiemy się tą drugą częścią.
To co to te mechanizmy w ogóle?
Jeżeli patrzymy na jakąkolwiek reakcję chemiczną, to zwykły rzut oka na substraty i produkty mówi nam tyle samo o tej reakcji, co zwykły rzut oka pod maską samochodu i to jeszcze w dodatku kiedy sami się na tym nie znamy i wiemy jedynie, gdzie wlewać płyn do spryskiwaczy. A więc słowem – mało nam to mówi!
Mechanizm natomiast, jest jakby rozebraniem całej reakcji na pojedyncze, najbardziej elementarne części, a więc niczym rozebranie silnika na pojedyncze części i śrubki, bo tylko tak nauczymy się w jaki sposób ten samochód właściwie jeździ.
Należy pamiętać, że mechanizm jest jedynie hipotezą – propozycją, którą później należy udowodnić eksperymentalnie!
Wyobraźmy sobie sumaryczną reakcję :
\(2O_{3} \rightarrow 3O_{2} \)
Do tej reakcji mamy dodatkowo mechanizm :
\((1) \ O_{3} \rightarrow O_{2} + O \)
\((2) \ O_{3} + O \rightarrow 2O_{2} \)
A więc reakcję sumaryczną rozłożyliśmy na dwa etapy ukazujące mechanizm reakcji. Pojawiło się nowe indywiduum : \(O \) , które definiujemy jako produkt pośredni – a więc taki związek, jon, rodnik, cokolwiek, co generalnie powstaje (reakcja 1), ale potem ostatecznie wchodzi w kolejną reakcję (reakcja 2) i takiego produktu pośredniego nie znajdziemy ani w produktach, ani w substratach reakcji sumarycznej, zatem łatwo taki produkt pośredni zidentyfikować (pomijając fakt, że jest to zazwyczaj jakaś ,,dziwna” cząsteczka tj. zwykle niestabilna, mocno reaktywna, np. jakiś karbokation, który pojawia się przy addycji HCl do propenu). Zerknijcie teraz na 55 edycja, II etap – Zadanie 3, podpunkt g)
Udowadniając to, co napisałem powyżej, po dodaniu stronami obu etapów reakcji (1) + (2) otrzymamy :
\((1) + (2) \Big ( O_{3} + O_{3} + O \rightarrow O_{2} + O + 2 O_{2} \Big ) \implies 2O_{3} \rightarrow 3O_{2} \)
I faktycznie, po skróceniu po obu stronach \(O \) otrzymujemy naszą sumaryczną reakcję. I widać, że po samym spojrzeniu na sumaryczną reakcję, trochę rzeczy może nam umknąć! Bo skąd mamy wtedy wiedzieć, że przez chwilę powstaje \(O \) ? A czy to w ogóle ważne, skoro ostatecznie nie występuje w sumarycznym równaniu?
Jest to bardzo ważne 🙂 !
Zanim przejdziemy dalej, chciałbym powiedzieć o cząsteczkowości reakcji – jest to po prostu liczba (cyfra) , która mówi nam o tym, ile cząsteczek bierze udział w danej reakcji. Na naszym przykładzie powyżej, widzimy, że w reakcji (1) jest tylko jedna cząsteczka, zatem reakcja jest jednocząsteczkowa (monomolekularna), natomiast w reakcji (2) biorą udział dwie cząsteczki, zatem mowa o reakcji dwucząsteczkowej (bimolekularnej). Są możliwe jeszcze reakcje trzycząsteczkowe (trójcząsteczkowe), ale są już one bardzo rzadkie. Natomiast reakcje czterocząsteczkowe nie są w ogóle znane!!!
Dlaczego? Musimy sobie odpowiedzieć na podstawowe pytanie – czym właściwie jest reakcja chemiczna? Z punktu widzenia kinetyki, będzie to tzw. zderzenie efektywne pomiędzy reagującymi ze sobą cząsteczkami. A zderzenie efektywne, to takie, podczas którego cząsteczki są odpowiednio zorientowane (wiązanie powstaje w konkretnym miejscu, a nie gdziekolwiek) i jeszcze w dodatku pędzą na siebie z odpowiednią prędkością (energią), bo gdy ta energia będzie niewystarczająca to one się po prostu od siebie odbiją. Więc zajście reakcji to nie jest jakaś trywialna sprawa! Zatem teraz spróbujcie sobie sami wyobrazić, jakie jest prawdopodobieństwo, że cztery cząsteczki (czyli reakcja czterocząsteczkowa) zderzą się ze sobą w tym samym czasie, z odpowiednią prędkością i w odpowiednim ułożeniu przestrzennym, żeby zaskutkowało to zderzeniem efektywnym i powstał jakiś tam produkt? Dlatego właśnie takich reakcji nie znamy i z tego wszystkiego wynika dla Was ważny wniosek! Jeżeli będzie trzeba zaproponować mechanizm, to żaden etap (żadna reakcja elementarna) nie może mieć w sobie więcej niż trzy cząsteczki, a właściwie to powinniście się trzymać maksymalnie dwóch cząsteczek, ze względu na to, że reakcje o cząsteczkowości równej 3 są bardzo rzadkie!
Kolejna ważna rzecz, na której bazujecie w wyznaczaniu wyrażenia na równanie kinetyczne danej reakcji (co niemalże zawsze idzie w połączeniu z przybliżeniem stanu stacjonarnego). Skoro daną reakcję mamy rozpisaną na etapy, czyli tzw. reakcje elementarne (które są niczym nasze śrubki w samochodzie), to w takich przypadkach jest dla nas bardzo sympatycznie, ponieważ równanie kinetyczne (w sensie odpowiednie rzędy reakcji ze względu na każdy substrat) dla takiej reakcji podstawowej (elementarnej) jest zgodne z cząsteczkowością tej reakcji!
Z pewnością z Atkinsa kojarzycie, kiedy zaczynaliście się w ogóle uczyć kinetyki, że dla przykładowej, pozornie prostej reakcji :
\(H_{2} + Br_{2} \rightarrow 2HBr \)
równanie kinetyczne na poziomie licealnym miałoby postać :
\(v = k[H_{2}][Br_{2}] \)
A więc prosta sprawa – szybkość reakcji zależy jedynie od produktów i wprost proporcjonalnie do stężeń/ciśnień substratów, podniesionych do potęgi równej współczynnikowi stechiometrycznemu (a więc np. dla reakcji \(2A \rightarrow P \) miałoby postać \(v = k[A]^{2} \) ). A potem w podręczniku okazuje sie, że równanie kinetyczne dla syntezy HBr (oczywiście nie uczymy się tego na pamięć 😀 ) wygląda następująco :
\(v = \frac{k[H_{2}][Br_{2}]^{\frac{3}{2}}}{[Br_{2}] + k'[HBr]} \)
A więc postać, której kompletnie się nie spodziewaliśmy! Co ciekawe, nie określamy tutaj całkowitego rzędu reakcji, ani też nie da się przypisać rzędu reakcji ze względu brom oraz kwas HBr (rzędy nieokreślone). Wniosek jest zatem taki, że faktycznie ,,nie mamy prawa” pisać sobie równania kinetycznego w oparciu o równanie sumaryczne, bo jak już wiemy, nic nam ono nie mówi o przebiegu reakcji (mechanizmie) = nie mamy pojęcia jak będzie wyglądać równanie kinetyczne.
Ale sytuacja, w której mamy już rozpisane wszystkie najprostsze, elementarne reakcje możemy już traktować według ,,schematu licealnego” , bo mamy przedstawioną daną reakcję elementarną na tacy, widzimy każdy produkt pośredni, wiemy dokładnie co oraz ile reaguje.
Oto trzy żelazne zasady, których należy się trzymać podczas układania mechanizmu reakcji :
- Suma reakcji elementarnych ma ostatecznie dać reakcję sumaryczną (tak jak u nas w przykładzie z ozonem, reakcja (1) oraz (2) czyli reakcje elementarne, po dodaniu do siebie stronami, a więc (substraty + substraty = produkty + produkty) dało po skróceniu reakcję przekształcania ozonu w tlen).
- Twoje rozpisane reakcje elementarne muszą być sensowne (co w praktyce oznacza raczej cząsteczkowość = 1 lub 2).
- To mechanizm ma się zgadzać z doświadczalnie wyznaczonym równaniem kinetycznym, a nie odwrotnie!
Tutaj zadanie można powiedzieć również z zakresu chemii organicznej, w którym należało zaproponować mechanizm reakcji 52 edycja, II etap, zadanie 3 : część b
Warto zwrócić uwagę na wykresy energetyczne opisujące reakcje, tutaj zajmiemy się reakcją najprostszego typu \(X \rightarrow Y \) :
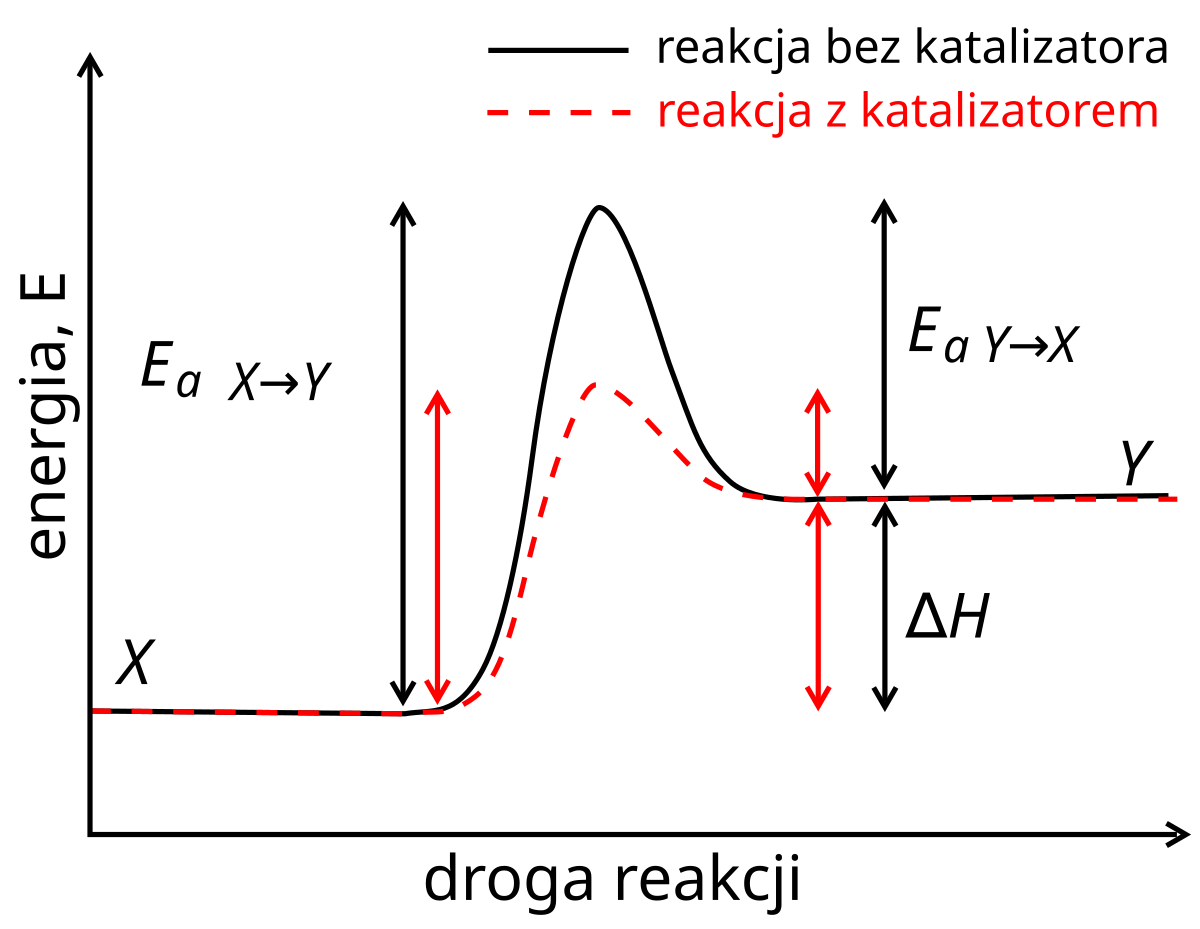 Pojawia nam się tutaj \(E_{a} \) co jest oczywiście energią aktywacji.
Pojawia nam się tutaj \(E_{a} \) co jest oczywiście energią aktywacji.
Energia aktywacji (\(E_{a} \) ) – można to uznać za minimalną energię (kinetyczną), która jest potrzebna, aby zaszła dana reakcja. Jeśli tej bariery nie uda się przekroczyć, to dana reakcja nie zajdzie.
Po lewej stronie wykresu mamy \(E_{a \ X \rightarrow Y} \) co oznacza tak naprawdę energię aktywacji dla reakcji ,,w prawo” , natomiast \(E_{a \ Y \rightarrow X} \) oznacza energię aktywacji dla reakcji ,,w lewo”. Zauważcie ważną rzecz (oj, może być takie zadanko!), że entalpia reakcji to różnica obu energii aktywacji a na tym wykresie będziemy mieć dodatnią wartość entalpii = reakcja endotermiczna \(\Delta H > 0 \) . Mam nadzieję, że idąc na II etap posiadacie podstawową wiedzę z zakresu równowagi chemicznej oraz termochemii, może to być np. wykorzystane w wydaniu kinetycznym chociażby w Równanie Eyringa . Sama równowaga to mam nadzieję, że nie trzeba tłumaczyć, motyw ten wykorzystałem w próbnym I etapie, patrz : Zadanie 3
Energia w maksymalnym punkcie tego wykresu obrazuje energię stanu przejściowego, a więc bardzo delikatnego stanu, w którym mamy takie samo prawdopodobieństwo, że reakcja pójdzie w prawo i w lewo.
Zwróćcie też uwagę na działanie katalizatora – sprawia on, że osiągnięcie tego najtrudniejszego dla cząsteczek momentu (a więc przełamanie energii aktywacji) jest łatwiejsze, ponieważ energia aktywacji się obniża (,,szybciej wejdziemy pod górę”)!
Ostatnia rzecz, o której chciałbym dzisiaj wspomnieć to mechanizm reakcji rodnikowych (wolnorodnikowych). Gdybym miał startować w tym roku, nauczyłbym się tego mechanizmu (w sensie ogólnych jego zasad), ale wybór zostawiam Wam, bo teoretycznie jeszcze czegoś takiego nie wymagano.
Reakcje rodnikowe są po prostu przykładem reakcji łańcuchowych, a więc takich, w których produkt pośredni generuje (tworzy) kolejny produkt pośredni itd. Wyróżniamy następujące fazy tej reakcji :
- inicjacja = skoro inicjacja alkoholowa to rozpoczęcie picia alkoholu, to inicjacja reakcji rodnikowej to etap, w którym rozpoczynamy tworzenie rodników.
- przykład \(Br_{2} \rightarrow 2Br \cdot \) Taka ,,reakcja” powinna Wam się wydawać dość nienaturalna (wymuszona) i faktycznie – żeby zaszła potrzebujemy ciepła (termoliza) bądź fotonów (fotoliza : \(h \nu \)).
- propagacja – propagowanie ,,teorii” , że szczepionki są złe i autyzm itd. to propagowanie głupoty = roznoszenie głupoty, zatem propagacja w reakcji rodnikowej polega na generowaniu kolejnych rodników (roznoszeniu rodników)
- przykład \(H_{2} + \cdot Br \rightarrow \cdot H + HBr \)
- przykład \(\cdot O \cdot + H_{2}O \rightarrow HO \cdot + \cdot OH \) *celowo rozpisałem dwie cząsteczki \(\cdot OH \) osobno, żeby zwrócić uwagę na fakt, że jeden rodnik generuje tutaj dwa rodniki.
- hamowanie – etap, który niekoniecznie musi występować, a wręcz nie chcemy, żeby występował, bo poza tym, że utrudnia wyprowadzenie równania kinetycznego, to jak z nazwy się domyślamy, zmniejsza szybkość reakcji
- przykład \(\cdot H + HBr \rightarrow H_{2} + Br \cdot \)
- w powyższym przykładzie (który dotyczy sumarycznej reakcji syntezy bromowodoru : \(H_{2} + Br_{2} \rightarrow 2HBr \) ) mamy sytuację, w której produkt (\(HBr \)) już sobie fajnie powstał, a tu nagle się zużywa na tworzenie kolejnych rodników, dlatego ten etap będziemy wyróżniać jako hamowanie, bo zmniejsza się stężenie produktu.
- inhibicja – nie ukrywam, że dość łatwo to pomylić z hamowaniem. Chodzi tutaj o to, że powstałe rodniki (nośniki, propagatory łańcucha) są usuwane w jakiś dziwny sposób, np. obijając się o ściankę naczynia (więc dla nas bez sensu, bo nie zaskutkuje to powstaniem produktu), lub gdy utracimy ten rodnik przez reakcję z obcym reagentem (można myśleć o inihibicji jako o reakcjach pasożytniczych, a więc nic nie wnoszących, a jedynie działających negatywnie).
- przykład \(\cdot H + HBr \rightarrow H_{2} + Br \cdot \)
- terminacja – nie możemy tak w nieskończoność tworzyć tych rodników, w końcu trzeba je ,,skasować” i to jest właśnie etap, który nazywa się terminacją.
- przykład \(A + \cdot X \rightarrow AX \)
Z pewnością chciałbym zwrócić jeszcze Waszą uwagę na mechanizm Lindemanna, może się to pojawić.
Jest on używany (nie jest to co prawda jedyna możliwość, bo tych mechanizmów imiennych jest jeszcze cała masa, ale jakby coś takiego miało się pojawić, to pewnie Lindemann) dla reakcje typu \(A \rightarrow P \) czyli substrat A przechodzi w jakiś produkt/y P. Wydawałoby się, że to jakaś reakcja jednocząsteczkowa, ale niekoniecznie tak musi być :
Sumarycznie :
\(A \rightarrow P \)
Mechanistycznie :
\((1) \ A + M \rightarrow A^{*} + M \) o stałych odpowiednio w przód i tył \(k_{1} , k_{2} \)
\((2) \ A^{*} \xrightarrow{k_{3}} P \)
M to może być dowolna cząsteczka (nawet taka sama jak A) i możemy sobie ją tak potraktować jako taką sprężynkę, która wzbudza energetycznie substrat A, co zapisujemy jako A*
Szybkość tworzenia produktu P wynosi : \(\frac{d[P]}{dt} = k_{3}[A^{*}] \)
Z przybliżenia stanu stacjonarnego można wyznaczyć stężenie produktu pośredniego :
\([A^{*}] = \frac{k_{1}[A][M]}{k_{2}[M] + k_{3}} \)
I podstawiając to do równania na szybkość tworzenia P :
\(\frac{d[P]}{dt} = \frac{k_{1}k_{3}[A][M]}{k_{2}[M] + k_{3}} \)
Przykłady do przećwiczenia wyprowadzania równań kinetycznych :
Wrzucam kilka zadań na przećwiczenie wyprowadzania równania kinetycznego na podstawie zaproponowanego mechanizmu reakcji (UWAGA – reakcje sumaryczną należy samemu zapisać!) :
Przykład 1 :
\((1) \ NO_{2} + F_{2} \xrightarrow{k_{1}} NO_{2}F + F \ (wolno) \)
\((2) \ NO_{2} + F \xrightarrow{k_{2}} NO_{2}F \ (szybko) \)
Odpowiedź : \(v = k_{1}[NO_{2}][F_{2}] \) lub gdy założyć, że \(k_{1} = k \) to otrzymujemy : \(v = k[NO_{2}][F_{2}] \)
Przykład 2 :
\((1) \ NO + O_{2} \leftrightarrows NO_{3} \ (szybko) \) , stałe odpowiednio w przód i w tył : \(k_{1} , k_{2} \)
\((2) \ NO_{3} + NO \xrightarrow{k_{3}} 2NO_{2} \ (wolno) \)
Odpowiedź : \(v = k[NO]^{2}[O_{2}] \) gdzie \(k = \frac{k_{1}k_{3}}{k_{2}} \)
*Jeżeli w tym zadaniu używałeś/aś przybliżenia stanu stacjonarnego, to powinno wyjść : \(v = \frac{k_{1}k_{3}[NO]^{2}[O_{2}]}{k_{2} + k_{3}[NO]} \) . Jakie wtedy należy przyjąć założenie ( i czy jest ono słuszne?), aby to równanie sprowadziło się do tej postaci powyżej?
Odpowiedź : \(k_{2} >> k_{3}[NO] \implies k_{2} + k_{3}[NO] \approx k_{2} \) . Warunek ten jest słuszny, ponieważ \(k_{2} >> k_{3} \) , bo pierwsza reakcja jest szybka, a druga wolna.
Przykład 3 (użyj przybliżenia stanu stacjonarnego) :
\((1) \ 2NO \leftrightarrows N_{2}O_{2} \ (szybko) \) , stałe odpowiednio w przód i w tył : \(k_{1} , k_{2} \)
\((2) \ N_{2}O_{2} + Cl_{2} \xrightarrow{k_{3}} 2NOCl \ (wolno) \)
Odpowiedź : \(v = \frac{1}{2} \frac{d[NOCl]}{[dt]} = \frac{k_{1}k_{3}[NO]^{2}[Cl_{2}]}{k_{2} + k_{3}[Cl_{2}]} \) , a zakładając, że \(k_{2} >> k_{3} \) otrzymamy ostatecznie \(v = \frac{1}{2} \frac{d[NOCl]}{[dt]} = \frac{k_{1}k_{3}}{k_{2}} [NO]^{2}[Cl_{2}] \)
Przykład 4 (załóż, że I etap jest w stanie równowagi, opisywany stałą K) :
\((1) \ NO_{2}Cl \leftrightarrows NO_{2} + Cl \ (szybko) \) , stałe odpowiednio w przód i w tył : \(k_{1} , k_{2} \)
\((2) \ NO_{2}Cl+ Cl \xrightarrow{k_{3}} NO_{2} + Cl_{2} \ (wolno) \)
Wskazówka : najłatwiej szybkość wyrazić za pomocą chloru tj. \(v = \frac{d[Cl_{2}]}{dt} \) . Zwróćcie uwagę na stechiometrię sumarycznej reakcji!! Bardzo podobny motyw był tutaj : 55 edycja, FW – 4B
Odpowiedź : \(v = – \frac{1}{2} \frac{d[NO_{2}Cl]}{dt} = 2 \frac{d[Cl_{2}]}{dt} = 2k_{3} \cdot K \frac{[NO_{2}Cl]^{2}}{[NO_{2}]} \) gdzie K to stała równowagi dla pierwszej reakcji.
Część druga niebawem (chyba, że nie wyrobie się z tworzeniem próbnego II etapu, wtedy zostawimy to na ostatni tydzień). Będzie ona dotyczyć kinetycznych aspektów reakcji organicznych.






8 komentarzy do “Mechanizmy w kinetyce chemicznej – cz.1”
Skąd się wzięło k3 we wzorze na stężenie stanu pośredniego ?
A który przykład?
W tekście a->p jak obliczasz a*
Jest to produkt pośredni i możemy ze względu na niego zastosować przybliżenie stanu stacjonarnego – odsyłam do Atkinsa.
W przykładzie 4 (równanie 2) chyba brakuje dwójki przed chlorem (drobny szczegół). Wydaje mi się, że w przykładzie 2 przedstawiony mechanizm utleniania tlenku azotu(II) jest inny niż w Atkinsie (w nim produktem pośrednim tej reakcji jest N2O2, a tu jest NO3 (jak przy rozkładzie tlenku azotu(V)), chyba, że tak też może być i jest to jakiś mechanizm alternatywny?
Bilans jest ok – bo tam jest NO2Cl w substratach, więc atomy chloru się zgadzają. Do jednej reakcji może być zaproponowanie wiele różnych ścieżek mechanistycznych, także na zawodach możesz dostać jeszcze inny mechanizm tej reakcji 🙂
Aha, to dzięki za wyjaśnienie, bo myślałem, że jest to jakoś udowodnione jak wyglądają produkty pośrednie danej reakcji jak np. w organicznej. Co do tej dwójki, to sorki, po prostu źle sobie do zeszytu polecenie przepisałem i nie sprawdziłem.
Nie ma problemu – w razie czego wciąż czekam na wiadomość na fb 🙂