


Mechanizmy w kinetyce chemicznej – cz. 2
Jeżeli ktoś jeszcze nie czytał, to polecam zacząć od tego postu : Mechanizmy w kinetyce chemicznej – cz.1
Dzisiaj zajmiemy się mechanizmami pod kątem chemii organicznej. Nie będzie to post o samym rysowaniu mechanizmów, jest to temat na zupełnie inną lekcję i to nie jedną, a kilka. Dzisiejszy post jest celowo krótki, bo doskonale zdaję sobie sprawę, że kilka dni przed zawodami nie ma czasu na czytanie, a przecież jeszcze tyle rzeczy trzeba powtórzyć!
Teraz natomiast postaram się nakreślić kilka zagadnień, które mogłyby zostać poruszone z okazji zadania pierwszego na II etapie, który już za dosłownie kilka dni! Za każdym razem ciężko uwierzyć, że to już minęło – nie żebym Was stresował. Na pewno jesteście już dobrze przygotowani i tylko dopinacie ostatnie szczegóły, robicie ostatnie powtórzenia 🙂
Ustalanie danego mechanizmu to często metoda prób i błędów. Owszem, do nieznanej nam reakcji jesteśmy w stanie zaproponować jakiś rozsądnie wyglądający mechanizm, jednak pamiętajmy, że kartka przyjmie wszystko i decydują wyniki doświadczalne. Można wykonać kilka prób, które wyeliminują bądź potwierdzą nasze przewidywania, przykładowo :
- przypuszczamy, że reakcja biegnie według mechanizmu rodnikowego – można wtedy dodać inicjator rodnikowy, który będzie przyspieszał tworzenie rodników, a zatem powinien przyspieszać całą reakcję (co już łatwo sprawdzić) lub też odwrotnie możemy dodać inhibitora rodników.
- przykładem inicjatora rodnikowego są (dwa najważniejsze przykłady, warto je po prostu znać na rzecz chemii organicznej po prostu) :
- azozwiązki – o wzorze ogólnym \(R-N=N-R \) z typowym przykładem w postaci AIBN czyli azobis(izobutyronitryl) o wzorze :

- reakcja tworzenia rodnika :
- nadtlenki organiczne – o wzorze ogólnym \(R-O-O-R \) z typowym przykładem w postaci nadtlenku benzoilu o wzorze :
- azozwiązki – o wzorze ogólnym \(R-N=N-R \) z typowym przykładem w postaci AIBN czyli azobis(izobutyronitryl) o wzorze :
- przykładem inicjatora rodnikowego są (dwa najważniejsze przykłady, warto je po prostu znać na rzecz chemii organicznej po prostu) :
- równanie kinetyczne – mówi nam o tym ile cząsteczek bierze udział w kluczowym czyli limitującym etapie reakcji.
- znakowanie izotopowe – przydaje się w sytuacji, kiedy chcemy prześledzić dokładny los danego atomu. Jak zobaczycie izotop w jakiejkolwiek reakcji to nie należy się tego bać, reakcja zazwyczaj przebiega zupełnie normalnie, po prostu w danej reakcji należy zwrócić uwagę, gdzie konkretnie dany atom wędruje i uważnie śledzić tą jego wędrówkę (co może się wiązać z podstawową znajomością mechanizmu, jak to było w reakcji hydrolizy estru na I etapie : 58 edycja, I etap, Zad 4a) . [Swoją drogą, myśleliśmy wtedy jako zawodnicy, że oto powoli do olimpiady wkraczają mechanizmy, ale jednak się myliliśmy. Teraz jestem ciekaw czy tegoroczne ,,nukleofile i elektrofile” w FW to zalążek wprowadzania mechanizmów...] Przykładowe zastosowanie? Chcemy się dowiedzieć, kto jest dawcą wodoru w danej reakcji czy powiedzmy rozpuszczalnik (woda) czy jakiś inny związek). Używamy zatem \(D_{2}O \) i sprawdzamy czy w produkcie końcowym jest atom deuteru. Jeśli go nie ma, to znaczy że woda nie jest dawcą wodoru w tym etapie.



Skoro już mowa o znakowaniu izotopowym to koniecznie należy powiedzieć o tzw. KIE czyli Kinetycznym Efekcie Izotopowym (było tutaj : 57, II, Zadanie 3) – definiujemy go jako zmiana szybkości reakcji, która jest spowodowana zamianą jednego atomu na jego izotop (czyli przykładowo atom wodoru zostaje zastąpiony atomem deuteru). Formalnie, oblicza się to poprzez podzielenie stałej szybkości reakcji z izotopem ,,lżejszym” przez stałą szybkości z izotopem ,,cięższym” o stałych szybkości, odpowiednio przyjmijmy \(k_{L} \) oraz \(k_{H} \) , a zatem :
\(KIE = \frac{k_{L}}{k_{H}} \)
Lub zakładając, że mamy atom wodoru oraz deuteru, moglibyśmy napisać :
\(KIE = \frac{k_{H}}{k_{D}} \)
*KIE jest najbardziej ,,wyrazisty, zmieniony” gdy różnica mas w obu izotopach jest duża. Zobaczcie, że różnica masy pomiędzy wodorem a deuterem jest aż dwukrotna, podczas gdy zmiana \(^{12}C \) na \(^{13}C \) jest już dużo mniejsza.
Typowo reakcja z cięższym izotopem przebiega wolniej, ponieważ potrzeba wówczas większego nakładu energetycznego aby osiągnąć stan przejściowy (łatwo to po prostu zapamiętać – ciężkim (,,grubym”) atomom ciężej doczłapać się do stanu przejściowego = wolniejsza reakcja = mniejsza wartość stałej) co typowo powoduje, że \(KIE > 1 \)
Najczęściej kinetycznego efektu izotopowego doświadczymy w formie pierwszorzędowej, co oznacza, że wiązanie pomiędzy danym atomem a wprowadzonym (i zastąpionym) izotopem jest zrywane bądź tworzone.
Na Olimpiadzie zdecydowanie mógłby pojawić się elementarny mechanizm \(S_{N}1 \ vs \ S_{N}2 \) , który każdy z Was powinien chociaż kojarzyć z chemii organicznej. Popatrzmy na tą samą reakcję, rozrysowaną według tych dwóch, diametralnie różnych mechanizmów :
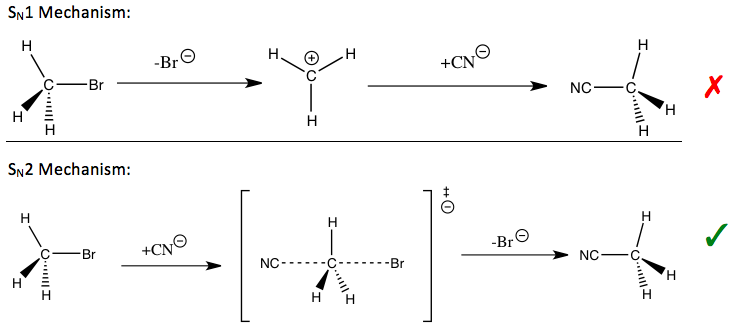
- reakcja \(S_{N}1 \) polega na wytworzeniu w pierwszej kolejności karbokationu, co oczywiście jest najbardziej ,,nienaturalnym” etapem tej reakcji, czyli limitującym. Karbokation bardzo szybko chciałby się pozbyć swojego ładunku, dlatego bez większej różnicy już mu, jaki nukleofil go zaatakuje (tutaj jest to grupa cyjankowa). Pierwszy etap zatem przebiega zdecydowanie najdłużej, a drugi w porównaniu z nim jest błyskawiczny.
- reakcja \(S_{N}2 \) jest reakcją jednoetapową, z wytworzeniem stanu przejściowego. Tutaj zatem ma już znaczenie rodzaj wybranego nukleofila. Zwróćcie też uwagę na wzajemną relację pomiędzy atakującym nukleofilem a grupą odchodzącą (opuszczającą czyli \(Br^{-} \) ) – są one pod kątem 180 stopni, co oczywiście jest logiczne, ponieważ ten brom musi mieć miejsce ,,na ucieczkę” . Ma to ogromne znaczenie w sytuacji, kiedy taka reakcja zachodzi na centrum stereogenicznym, bo dojdzie wówczas do tak zwanej inwersji konfiguracji (czyli jej zmianie, np. z \(R \rightarrow S \) lub z \(S \rightarrow R \)) , co nosi też nazwę inwersji Waldena.
W powyższej reakcji najlepszą opcją wydaje się oznakowanie węgla np. na izotop \(^{13}C \) w cząsteczce bromometanu, ponieważ to na tym węglu reakcja się ,,dzieje”.
Dla tej reakcji kinetyczny efekt izotopowy (może to być w zadaniu podane lub też do wyliczenia – zobaczcie koniecznie to zadanie : 57 edycja, II etap – Zadanie 3 )wynosi :
\(KIE = \frac{k_{^{12}C}}{k_{^{13}C}} \approx 1,09 \)
Jak to interpretować? Jest to bardzo mała wartość, pokazująca, że podstawienie izotopem węgla właściwie nie zmieniło szybkości reakcji. Zobaczcie, że gdyby reakcja biegła według mechanizmu \(S_{N}1 \) , w którym podczas etapu limitujacego reakcję (pierwszego etapu) uczestniczy nasz węgiel, który potem zamieniliśmy na izotop, to różnica w szybkości reakcji byłaby dość znaczna, przykładowo KIE = 4
W ten sposób udowadniamy, że reakcja biegnie jednak według mechanizmu \(S_{N}2 \) , co mam nadzieję, sami byście po prostu wiedzieli w tym przypadku, jako że konkurencyjny mechanizm zakłada tworzenie fatalnego karbokationu.
I na koniec tych mechanistycznych przygód, zadanie z 61 edycji – 61, II – 4 (tamtejsze dwa zadania z organicznej to było istne piekło! Kto zrobił je na >25 pkt, to jest naprawdę kozak!) :

Mamy ustalić strukturę produktu A oraz wybrać odpowiedni produkt pośredni, który tworzy się w tej reakcji.
Mamy podaną masę molową, którą wystarczy porównać z wyjściowym alkinem i niełatwo wydedukować, że wystarczy dodać 3 atomy deuteru i mamy produkt. Pozostaje pytanie, który produkt pośredni wybrać?
Tak na chłopski rozum, to produkty pośrednie 1 oraz 3 nie różnią się niczym, a przede wszystkim, nie ma tam żadnej zmiany w obrębie alkinu, który jest naszym centrum reakcji. W produkcie 2 mamy dodatkowo podstawiony atom miedzi za wodór, co mogłoby tłumaczyć, dlaczego oba atomy przy końcu terminalnym nowo powstałego alkenu są atomami deuteru (bo sumarycznie dodajemy trzy atomy deuteru, a zatem niesymetrycznie, zatem 2 atomy deuteru dodamy symetrycznie po jednej i drugiej stornie, pytanie co zrobić z trzecim? Dodajemy go na terminalnym końcu, bo tam jest ta miedź.
Na zakończenie polecam jeszcze zrobić to zadanie : 52 edycja, II etap – Zadanie 3 część B






6 komentarzy do “Mechanizmy w kinetyce chemicznej – cz. 2”
Ciężko zdać sobie sprawę, że za tydzień będzie już po wszystkim, a w następnym tygodniu czeka mnie jeszcze sporo pracy
Ostatni tydzień jest wybitnie ciężki, chociaż przy okazji jest niepełny, byle do soboty 🙂 A potem należny odpoczynek!
Życzę sobie i wszystkim następnym pokoleniom, aby mechanizmy były wymagane tylko w postaci 52 edycji II.3 (podany przepis ogólny na chemii fizycznej i zrób coś z tym), ale nigdy na chemii organicznej jako: zakwalifikuj reakcje do E1, Sn1, Sn2, rozrysuj karbokation, czy może jak w McMurrym zaproponuj mechanizm. Oddzielmy liceum od studiów.
Hmm.. kontrowersyjny temat trzeba przyznać. Ja jestem zdania, że bez nauki i zrozumienia mechanizmów uczeń nie jest w stanie zapamiętać reakcji z Murry’ego. Taki jeden ogólny schemat substytucji nukleofilowej w obrębie grupy karbonylowej tłumaczy prawie jeden cały tom, w tym bardzo trudne reakcje związane z chemią enoli. Myślę, że niekoniecznie powinno się to pojawiać na Olimpiadzie pod szyldem ,,narysuj mechanizm” , ale jednak z wykorzystaniem jego elementów jako tłumaczenie powstaniawania np. konkretnego izomeru czy też stereochemii itd. To jest też przy okazji piękno Claydena, który nie przykładowo nie dyktuje reguły Markownikowa a po prostu ją tłumaczy!
Poza tym w konkursach chodzi właśnie o to, żeby wskoczyć o poziom wyżej, tak jak konkurs gimnazjalne były na poziomie matury podstawowej, tak samo tutaj powinny być elementy ze studiów 🙂
Rzeczywiście niektóre mechanizmy są warte poznania dla lepszego zrozumienia, ale dla mnie większość z nich zabiera tylko dane na moim neurodysku. Tymi studiami mnie Pan rozbawił, jako że statystyczny student chemii nie przeszedłby II etapu. Wydaje mi się, że organiczna stoi na tak wysokim poziomie, że nie ma potrzeby dodatkowego różnicowania uczniów przez wymaganie znajomości mechanizmów. Już bez tego można się spocić.
To prawda, zadania są bardzo trudne, ale raczej ich ciężkość leży na mocnej dedukcji a nie ogromnej wiedzy. Studia jak to studia, opierają się na beznamiętnym zapamiętywaniu i robieniu giełd, zamiast nauki, która ma jakiś sens. Coś jednak jest w tym, że jakbyś popytał zawodników z top 50 to oni wszyscy te podstawowe mechanizmy znają, a popytasz tych z top 250 i już różnie bywa. Więc być może jednak warto tych elementarnych się nauczyć. W Murrym nie jest to rewelacyjnie wytłumaczone, ale jednak Clayden robi na tym polu rewelacyjną robotę. Zobacz, że nauczywszy się np. mechanizmu addycji elektrofilowej poradzisz sobie bez problemu chociażby z addycją reagenta typu NOCl (53 edycja, II etap), podczas gdy dla kogoś, kto uczy się reakcji na pamięć będzie to problemem, bo po prostu takiej sytuacji nie widział, nie zna. Podejściem Claydenowskim nie uczysz się reguł (taki Markownikow jest tam podany w ramce Ciekawostka) więc odchodzi Ci zapamiętywanie takich regułek na neurodysku 😀